
Non-invasive Prenatal Testing for pregnancy as early as 10 weeks
Safe, Accurate, and Easy
Enrolling for Cordlife NIPT
Cordlife NIPT must be prescribed by your obstetrician who will explain the details about NIPT to ensure the test is suitable for you.
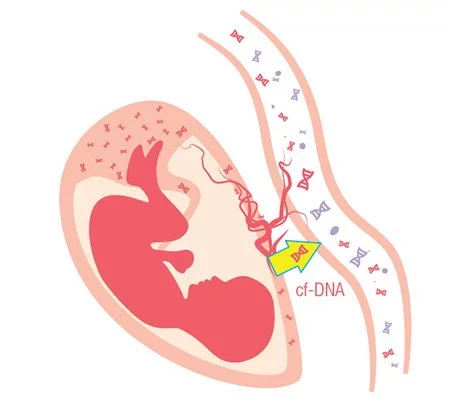
What is Non-Invasive Prenatal Test (NIPT)
Non-Invasive Prenatal Test (NIPT) is a blood test as early as 10 weeks of pregnancy to screen for common chromosomal abnormalities.
In 1997, scientists first reported the presence of small amounts of baby’s DNA (known as cell-free DNA, or cfDNA) in the mother’s blood as early as four gestational weeks1 . The rapid development of next generation sequencing technology makes it possible to detect the risk of having chromosomal abnormalities including Down Syndrome, Patau Syndrome, Edwards Syndrome and other genetic conditions non-invasively.
The American Congress of Obstetricians and Gynecologists (ACOG) Recommends all pregnant women regardless of age to be offered screening for foetal chromosomal abnormalities.2
What are the NIPT brands available?

From a simple blood draw as early as 10 weeks into your pregnancy, Cordlife NIPT screens for the most common chromosomal abnormalities that can affect your developing baby’s future. This test is done with little or no risk to your pregnancy.
Why Choose Non-Invasive Prenatal Testing?

Safe and Non-invasive
Only 10mL of mother’s blood will be drawn to screen for chromosomal abnormalities without harming the baby.

Early Screening
The test can be done as early as 10 weeks of pregnancy.

Screen chromosome abnormalities
Various screening panels available to detect genetic conditions.

Quality Testing
Your blood sample will be processed by world-class laboratory.

Most comprehensive technology
Uses Whole Genome Sequencing approach that reads millions of gene sequences to ensure comprehensive coverage with more than 99% accuracy.

Clear and Simple Report
Delivers critical information as definitive results that don’t rely on ambiguous numerical risk scores.
What does Non-Invasive Prenatal Test (NIPT) screen for?
Trisomies
T21 (Down Syndrome)
Affects one in 800 newborns
Individuals with Down Syndrome (Trisomy 21) are characterised by typical facial features such as an oval-shaped face and eyes that slant upwards. Most individual diagnose with Down syndrome will have some degree of learning difficulty, delayed speech development and a delay in motor development.
Approximately 50% of babies with Down syndrome are born with a heart defect and are also at risk of developing other medical conditions.
Although women of any age can have a child with Down syndrome, the chance of having a child with this condition increases with maternal age.
T18 (Edwards Syndrome)
Affects one in 5,000 newborns
Edwards Syndrome (Trisomy 18) is due to an extra copy of chromosome 18, which is associated with a high rate of miscarriages. Many individuals with trisomy 18 die before birth or within their first month. Only 5-10% of affected children live past their first year, and often have severe intellectual disability or congenital heart defects.
T13 (Patau Syndrome)
Affects one in 16,000 newborns
Patau Syndrome (Trisomy 13) is due to an extra copy of chromosome 13, which is associated with a high rate of miscarriages. Many foetuses don’t survive until full-term and are stillborn or spontaneously abort. Infants born with Trisomy 13 usually have severe congenital heart defects and other medical conditions, and survival beyond the first year is rare. Features include slow growth before birth, low birth weight, heart defects, organ malformation, brain and central nervous system abnormalities and craniofacial abnormalities.
T22 Trisomy 22
This disorder is found in individuals with an extra copy or a variation of chromosome 22 in some or all cells of their body. (Complete) Trisomy 22 is a frequent cause of spontaneous abortion during the first trimester of pregnancy, leading to the second most common cause of miscarriage after trisomy 16. Progression to the second trimester and livebirth are rare due to severe organ malformations associated with this condition.
Features associated with Trisomy 22 typically include growth delays, mental retardation, unequal development of the two sides of the body (hemidystrophy), and webbing of the neck.
T16 Trisomy 16
Trisomy 16 is a rare chromosomal abnormality in which there are three copies of chromosome 16 rather than two. Trisomy 16 is incompatible with life and nearly all affected babies are miscarried in the first trimester. As such, it is the most common trisomy leading to miscarriage.
T9 Trisomy 9
Trisomy 9 is a rare trisomy affecting the ninth chromosome either as full trisomy, trisomy mosaic or partial trisomy. Full trisomy 9 is always fatal; most babies with full trisomy 9 are miscarried in the first trimester. Those that make it to birth typically will not survive more than a few months, with most dying in the first week of life. Partial trisomy 9 and mosaic trisomy 9 have a more uncertain prognosis. Many babies with mosaic trisomy 9 die in infancy and those that survive usually have severe developmental impairments such as structural malformation to the heart (i.e. congenital heart defect). Partial trisomy 9 may not affect the baby’s life expectancy, but affected babies may have a range of common health and developmental problems.
Sex chromosome Aneuploidies
Trisomy X (Triple X Syndrome)
Affects one in 1,000 females
Triple X syndrome results from an extra copy of the X chromosome in each of a female’s cells. Females with this condition may be taller than average, but it typically causes no unusual physical features. Most females have normal sexual development and are able to conceive children. There is an associated risk of learning disabilities and delayed development of speech, language and motor skills. Seizures or kidney abnormalities occur in about 10% of affected females.
Monosomy X (Turner Syndrome)
Affects one in 2,500 females
Most often, Turner syndrome results when one X chromosome is normal and the other X chromosome is missing or altered.The most common feature of Turner syndrome is short stature, which becomes evident by about age 5. An early loss of ovarian function is also very common. Up to half of affected females are born with a heart defect, complications of which can be life threatening. However, most girls and women with the condition have normal intelligence.
XXY (Klinefelter Syndrome)
Affects one in 500 to 1,000 males
Most often, Klinefelter syndrome is the result of at least one extra copy of the X chromosome in each cell. The condition interferes with sexual development, such as reduced levels of testosterone, which can lead to delayed, or incomplete puberty, breast enlargement, reduced facial and body hair, and infertility. It also causes abnormal functioning of the testes.
XYY (Jacobs Syndrome)
Affects one in 1,000 males
Jacob’s syndrome is the result of one extra copy of the Y chromosome in each cell. Although males with this condition may be taller than average, this chromosomal change typically causes no unusual physical features. Most have normal sexual development and are able to father children.There is an increased risk of learning disabilities and delayed development of speech, language and motor skills. A small percentage of males are diagnosed with autistic spectrum disorders, which are developmental conditions that affect communication and social interaction.
Deletion Syndromes
5p deletion syndrome
(Cri du Chat Syndrome)
Affects one in 20,000 to 50,000 newborns
This condition results when a piece of chromosome 5 is missing. Affected infants have a high-pitched cry that sounds like a cat, hence the name Cri-du-chat (cat’s cry).It causes intellectual disability and delayed development, small head size, low birth weight, and weak muscle tone in infancy. Affected individuals also have distinctive facial features, including widely set eyes, low-set ears, a small jaw and a rounded face. Some infants with Cri-du-chat syndrome are born with a heart defect. As they grow, people with cri-du-chat usually have difficulty walking and talking. They may have behavioural problems and severe intellectual disability.
1p36 deletion syndrome
Affects one in 5,000 to 10,000 newborns
This condition results when a piece of chromosome 1 is missing, and typically causes severe intellectual disability. Most affected individuals have limited or no speech, as well as behaviour problems. Most have structural abnormalities of the brain resulting in seizures. There may also be weak muscle tone and swallowing difficulties. Physical characteristics include a small head that is also unusually short and wide in proportion to its size. There might also be vision or hearing problems, and abnormalities of the skeleton, heart, gastrointestinal system and other organs.
2q33.1 microdeletion syndrome
This rare condition results when a piece of chromosome 2 is missing. It affects the motor neurons, which are the specialised nerve cells in the brain and spinal cord that control the movement of muscles. The disorder usually causes severe mental retardation and behaviour problems.
16p12.2 deletion syndrome
This condition results when a piece of chromosome 16 is missing. Common clinical features for 16p12.2 deletion includes: a delay in starting to speak, a degree of developmental delay or learning difficulty, growth impairment, a cardiac malformation, epilepsy and mild dysmorphic facial features without a consistent pattern.
11q23 microdeletion syndrome
(Jacobsen Syndrome)
Affects one in 100,000 newborns
Jacobsen syndrome, which is also known as 11q terminal deletion disorder, is caused by a deletion of genetic material at the long (q) arm of chromosome 11. Most affected individual have delayed development in motor skills and speech, with most having cognitive and learning difficulties and distinctive facial features such as small/low set ears, widely set eyes and large head size. More than 90% of people with Jacobsen syndrome have bleeding disorder call Paris-Trousseau syndrome that causes lifelong risk of abnormal bleeding and easy bruising.
1q32.2 microdeletion syndrome
(Van der Woude Syndrome)
Affects one in 35,000 to 100,000 newborns
Van de Woude syndrome is a condition that affects the development of the face. Many people with this disorder are born with a cleft lip, a cleft palate (an opening in the roof of the mouth), and have an increased risk of delayed language development, learning disabilities, or other mild cognitive problems. Van der Woude syndrome is usually caused by mutations in the interferon regulatory factor 6 (IRF6) gene which is typically encoded in region on chromosomes
15q11.2 microdeletion syndrome
(Prader Willi / Angelman Syndrome)
Angelman syndrome affects one in 12,000 newborns whereas Prader Willi syndrome affects one in 20,000 to 50,000 newborns
Most mothers carrying babies with a 15q11.2 microdeletion experienced no pregnancy problems, had a normal delivery and only discovered their baby was affected after the birth. There are two main types of syndrome associated with this microdeletion. Angelman syndrome results from a loss of gene activity (expression). Main characteristics of Angelman syndrome includes severe mental retardation, lack of speech, excessive happy demeanour.
Prader Willi syndrome is caused by a loss of active genes in a region of chromosome 15. People can have either Prader-Willi syndrome or Angelman syndrome. Characteristics of Prader-Willi syndrome includes mild to moderate development delay, delayed to no puberty.
Legend:
Aneuploidy: a condition in which a person has one or a few chromosomes above or below the normal chromosome number (46). It originates during meiosis (a cell division process that gives rise to the sperm and egg) when the chromosomes do not separate properly between the two cells. For example, a common form of aneuploidy is when an individual have 3 copies of chromosome 21, which is more commonly known as Down syndrome.
How do I enroll for NIPT?
1. Inquiry
Inquire with your obstetrician about NIPT or send us an email with your full name, contact number, name of OB-GYN, and gestational age.
2. Blood drawn
Upon or after assessment by your obstetrician, a simple and non-invasive blood extraction will be done. Blood sample will be collected from the mother’s arm.
3. Sample processing
Your blood sample is analyzed by a Whole Genome Sequencing approach using Next Generation Sequencing Technology. Between 5 -10 million DNA fragments from the whole genome are read and sequenced to achieve the highest accuracy.
4. Report
Your doctor will receive your detailed report with a definitive result of “screen positive” or “screen negative”.
If the screening result is negative, it suggests a low-risk for the tested genetic conditions. Otherwise, women who screened positive for a particular chromosomal abnormality are advised to consult their obstetrician / doctor for further action, which may include a diagnostic confirmatory test, such as Amniocentesis or Chorionic Villus Sampling (CVS).
References:
- Jiang F, et al. BMC Medical Genomics, 2012 Dec;5:57. doi:10.1186/1755-8794-5-57
- Ob-Gyns Release Revised Recommendations on Screening and Testing for Genetic Disorders. American Congress of Obstetricians and Gynecologists.
https://www.acog.org/About-ACOG/News-Room/News-Releases/2016/Ob-Gyns-Release-Revised-Recommendations-on-Screening-and-Testing-for-Genetic-Disorders. Last accessed 22 October 2017. - Noninvasive prenatal screening for fetal aneuploidy, 2016 update: a position statement of the American College of Medical Genetics and Genomics. American College of Medical Genetics and Genomics.https://www.acmg.net/docs/NIPS_AOP.pdf. Last accessed on 16 October 2017
Cordlife NIPT Packages
Cordlife NIPT must be prescribed by your obstetrician who will explain the details about NIPT to ensure the test is suitable for you.
Disclaimer
No test is perfect. DNA test results do not provide a definitive genetic risk in all individuals. Cell-free DNA does not replace the accuracy and precision of prenatal diagnosis with CVS or amniocentesis. Patients with positive test result or an additional finding should be referred for genetic counselling and offered invasive prenatal diagnosis for confirmation of test results. A negative test result does not ensure an unaffected pregnancy. The absence of an additional finding does not indicate a negative result. While results of this testing is highly accurate, not all chromosomal abnormalities may be detected due to placental, maternal or fetal mosaicism, or other causes. The healthcare provider is responsible for the use of this information in the management of their patient.
NIPT is a screening, not a diagnostic test. Patients should opt for it only as an informed choice after appropriate pre-test counselling.
What our customers are saying




FAQs About Cordlife NIPT
Learn more about the most of the commonly asked questions about Cordlife NIPT.